一、前言
医疗器械安全性和有效性评价研究应采用科学、合理的评价方法,其中动物实验是重要手段之一,其属于产品设计开发中的重要研究,可为产品设计定型提供相应的证据支持;若需开展临床试验,可为医疗器械能否用于人体研究提供支持,降低临床试验受试者及使用者的风险以及为临床试验设计提供参考。
但并不是所有医疗器械均需要通过动物实验验证产品安全性和有效性。为了对开展动物实验的必要性判定提供指导,特制订本原则。
本原则为医疗器械动物实验研究技术审查指导原则系列中的第一部分,为判定是否开展医疗器械动物实验的决策原则,关于动物实验设计等其他方面的内容请参见其他部分指导原则。
本原则是供申请人和技术审评人员使用的技术指导性文件,不涉及注册审批等行政事项,亦不作为法规强制执行,如有能够满足法规要求的其他方法,也可以采用,但应提供充分的研究资料和验证资料。应在遵循相关法规的前提下使用本原则。
本原则是在现行法规和标准体系以及当前认知水平下制订的,随着法规和标准的不断完善,以及科学技术的不断发展,本原则相关内容也将进行适时地调整。
二、适用范围
本原则适用于决策医疗器械是否需在活体动物上进行在体实验,不包括在非活体动物、离体组织或器官上进行的研究。
以下情况可参考本原则:
(一)医疗器械申请人在设计开发阶段确定是否需要开展动物实验时;
(二)医疗器械监管机构在技术审评环节评价开展动物实验的必要性时。
本原则不替代GB/T16886系列标准等医疗器械生物学评价相关的技术文件。如通过动物实验方式评价医疗器械的生物相容性,亦应符合GB/T16886系列标准等生物学评价相关技术文件。
如有针对特定产品的指导原则发布,则遵循相应产品的指导原则。
本原则不适用于按照医疗器械管理的体外诊断试剂。
医疗器械临床试验伦理审查时,可参考本原则中适用部分以评估临床前动物实验的必要性。
三、基本决策原则
在医疗器械设计开发阶段,决策是否开展动物实验时,建议考虑动物福利伦理原则及风险管理原则。
(一)动物福利伦理原则
申请人需遵循动物实验的“替代(Replacement)、减少(Reduction)和优化(Refinement)”原则,即3R原则。
申请人在决策是否开展动物实验前,需要特别考虑动物福利伦理,充分开展实验室研究,不宜采用动物实验替代实验室研究。
若有经过确认/验证的非活体研究、计算机模拟等方法,则优先采用上述方法以替代动物实验。
申请人宜充分利用已有的信息获取产品安全性、有效性和可行性的相关证据,如可利用已有的同类产品动物实验数据或通过与市售同类产品进行性能比对等方式验证产品的安全性、有效性和可行性。若相关证据充分,可免于动物实验。
(二)风险管理原则
申请人在医疗器械设计开发时应进行充分的风险管理活动。风险控制作为风险管理的重要部分,是将风险降低并维持在规定水平的过程。实施每一项风险控制措施后应对其有效性予以验证(其中包括确认活动)。实验室研究或动物实验等均是验证风险控制措施有效性的手段,申请人宜尽可能地通过前期研究(如实验室研究等)对已识别风险的控制措施有效性进行验证,只有在实验室研究不足时,才考虑通过动物实验开展进一步验证。动物实验资料可作为风险/受益分析时的支持性资料。
如需通过动物实验进行风险控制措施有效性的验证,则结合动物实验目的,一般从可行性、有效性、安全性三方面进行考虑:
1.可行性
可行性研究是指产品设计开发阶段进行的,对产品工作原理、作用机理、设计、可操作性、功能性、安全性等方面进行确认/验证,或识别新的非预期风险的研究,如生物可吸收支架平台材料的筛选、经导管瓣膜置换装置的设计可行性、迭代设计更新的验证等。
2.有效性
尽管动物与人体之间,在部分医疗器械的有效性方面可能存在一定差异,但设计合理的动物实验可支持产品的有效性(包括性能和操作),如可吸收防粘连医疗器械的防粘连性能评价,组织修复材料引导组织重建的有效性评价,多孔涂层关节类产品或3D打印多孔结构产品的骨结合效果评价等。
3.安全性
申请人采取风险控制措施后,部分产品安全性可适当采用动物实验研究进行评价,如含药医疗器械中药物安全性范围研究,通过组织病理学等方式的毒理学评价、产品对生物体的损伤评价,动物源性材料的抗钙化性能,外科血管闭合设备的血管热损伤研究,防粘连器械与组织粘连相关并发症的评价等。
实验目的有时是不能严格划分界限的,因此一项动物实验可能同时对产品的可行性、有效性、安全性进行评价。
若产品采用新的作用机理、工作原理、设计、主要材料/配方、应用方法(如手术操作)、预期用途、增加新的适用范围、改进某方面性能等,申请人应针对产品创新点相关风险进行评估,并考虑通过动物实验对风险控制措施的有效性进行验证。
申请人可参考以下决策流程图进行是否开展动物实验的决策。
四、是否开展动物实验的决策案例
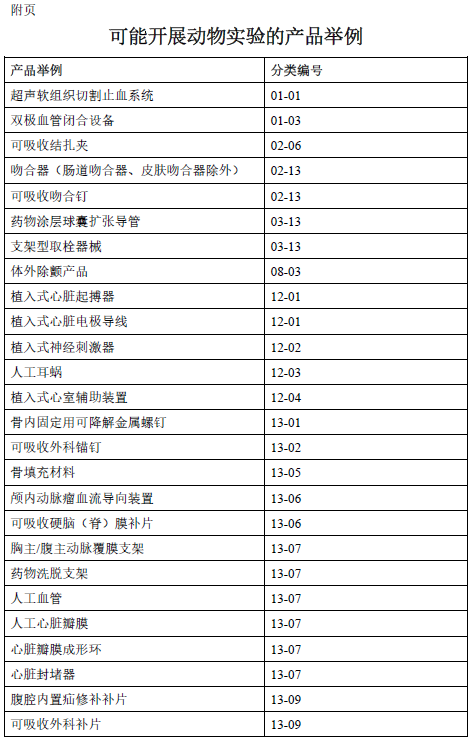
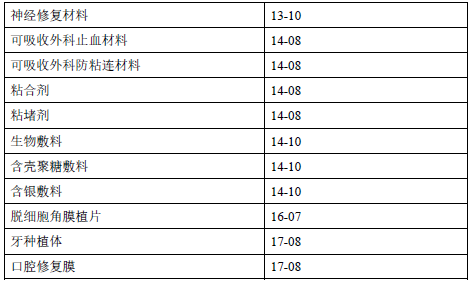
附页列出了部分可能需要开展动物实验的产品示例,需要注意的是,在不同的具体情形下,根据决策原则对于同样的产品可能将得出不同的判定结果;另外,对于表中未列的医疗器械产品,在某些具体情形下也可能需要开展动物实验。建议申请人根据产品实际情况参照决策流程图予以判定。
为便于理解决策原则,本章节列举了以下实际案例。相关案例仅针对特定产品在具体情形下的决策判定,例如同一申请人在前代植入式心脏起搏器基础上对产品功能的改进或更新,再例如采用新材料制造的肠道吻合器等。
(一)多孔涂层生物型髋关节假体
多孔涂层生物型髋关节假体的主要风险包括产品骨结合效果欠佳或涂层剥落造成的假体固定失败等,通过动物实验可评价涂层的骨结合效果。如果通过涂层的成分表征、形貌及体视学数据(厚度、孔隙率、孔隙尺度等)、涂层机械性能评价(涂层与基体结合强度等)、涂层稳定性及耐腐蚀性能评价、生物相容性评价等研究证明其与已上市同类产品的涂层具有等同性,则无需通过动物实验来评估多孔涂层的骨结合效果和涂层的稳定性。
(二)心电图机
心电图机的主要风险之一是工作数据的不准确性,包括心电图自动测量的不准确性和心电图自动诊断的不准确性。可进行实验室研究,通过心电图标准数据库来验证心电图自动测量的准确性,可通过形态诊断用心电图数据库和节律诊断用心电图数据库来确认公开形态解释的准确性和公开节律诊断的准确性,无需开展动物实验。
(三)注射用交联透明质酸钠凝胶
交联透明质酸钠凝胶可用于面部注射以纠正鼻唇沟皱纹,纠正效果一般可达到6个月。鉴于通过动物实验无法考察人体面部皱纹的改善程度,故一般不采用动物实验数据支持该类产品的有效性,建议在人体临床评价资料中关注产品注射6个月时的鼻唇沟皱纹严重程度分级(如WSRS)较术前的改善程度等疗效评价指标。
(四)可吸收生物疝修补补片
本案例提及的可吸收生物疝修补补片用于修复腹壁疝及腹壁缺损,一般具有类似细胞外基质的微观结构。产品植入人体后,宿主细胞在材料中生长,最终重塑出的腹壁组织对缺损进行修补。
1.该类产品最主要的风险之一在于疝或腹壁缺损的复发,宜采取一系列风险控制措施确保产品组织重建的有效性,以降低疝复发的风险。对于该类产品,仅仅依靠常规的实验室研究并不能验证疝复发风险相关控制措施的有效性,宜考虑使用组织病理学等动物实验数据验证组织重建效果。
2.申请人在开展动物实验前可收集已有同类产品的动物实验资料或文献数据,并分析这些数据能否用于支持申报产品组织重建效果的评价,如现有资料充分则无需开展动物实验。
(五)体外除颤产品
体外除颤产品供不同的使用者和操作者在不同的预期使用环境下进行体外电复律治疗。
对于该类产品,常规的实验室研究并不能验证体外电复律技术风险相关控制措施的有效性,因此宜使用活体动物开展实验获得除颤研究数据进行验证。
(六)超声软组织切割止血系统
超声软组织切割止血系统用于软组织切割和血管闭合产品通过摩擦产生的热量导致组织凝固后被切开、封闭血管(本例子不包含3mm以上血管的切割和闭合功能的特殊要求)。
1.该产品主要风险包括产品设计不合理等原因可能造成的血管切割闭合不充分和组织热损伤等。仅依靠实验室研究无法充分验证这些风险的控制措施是否有效,需要通过急性动物实验观察产品的即刻的血管切割闭合情况和组织热损伤情况,需要通过慢性动物实验观察热损伤愈合情况和继发出血情况,进而验证风险控制措施的有效性。
2.若申请的产品包含多个类似设计的刀头,可在开展动物试验前通过体外爆破压力实验筛选出性能最差的刀头开展动物实验,以起到减免部分动物实验的目的。对于新增与已有刀头相似(设计类似、性能相近)的刀头,可使用体外爆破压力实验结果证明其与相似刀头的等同性,不再开展动物实验。
(七)植入式心脏起搏器
1.植入式心脏起搏器属于高风险植入器械,开展动物实验可以为产品设计定型提供相应的证据支持。若同一申请人在前代产品基础上进行植入式心脏起搏器的改进或更新,对于前代产品已验证的内容无需开展动物实验,必要时申请人仅针对改进或更新部分开展相应的动物实验。
2.患者在植入心脏起搏器后一般不能进行核磁共振检查(MRI),如果申请人设计开发了MRI兼容的植入式心脏起搏器,需要评估MRI环境对产品安全性及有效性带来的影响,进行MRI兼容性相关研究。MRI兼容性研究通常需要应用动物进行计算机建模验证MRI兼容的安全性与有效性,当验证过计算机建模的准确性后,对于同一申请人其他植入式心脏起搏器产品的MRI兼容性研究,可以不再重复进行动物实验。
3.无导线起搏器与传统的植入式心脏起搏器相比,采用了新的结构设计、手术操作方法,不需要植入传统的植入式心脏电极导线,申请人宜针对创新点相关风险进行评估,并对风险控制措施有效性进行验证或确认,申请人宜对无导线起搏器开展动物试验,验证产品安全性、有效性及可行性。
(八)药物洗脱支架
1.尽管药物洗脱支架产品所含药物如紫杉醇、雷帕霉素已作为药品具有较长的临床应用历史,但在器械中应用与其单独作为药品应用时具有较大差异,例如药物洗脱支架植入后,靶血管壁中的局部组织药物浓度会远远高于药品系统使用后的血药浓度,单纯作为药品使用的药代及毒理学研究资料并不足以支持其安全性,宜进一步通过动物实验开展靶血管、远端心肌等局部组织的毒理学安全性研究,获取必要的组织病理学数据等。对于两个含有相同药物的药物洗脱支架,若支架药物载体材料不同,临床应用时同样存在较大差异,例如药物成分从不同载体材料中释放、吸收的速率不同,宜通过动物实验研究结合已有的文献数据资料来确认药物剂量密度及安全范围。
2.对于涂有可降解涂层的药物洗脱支架产品,其降解性能是载体聚合物材料筛选中一项重要因素。通过动物实验在体研究药物洗脱支架涂层的降解性能十分必要,但如果载体聚合物(例如聚乳酸-羟基乙酸共聚物,PLGA)的降解性能,可通过同类产品信息、文献数据信息、材料数据库信息、监管机构备案信息等获得支持,申请人无需对申报产品重新开展降解性能的动物实验。
(九)骨内固定用可降解金属螺钉
骨内固定用可降解金属螺钉,该类产品在骨愈合初期提供初始的稳定固定,待骨愈合后逐渐降解,避免二次手术取出。
该类产品主要风险包括降解周期与骨愈合周期不匹配造成内固定过早失效,以及降解产物对机体组织和器官带来的安全性问题等。对于该类产品,仅仅依靠常规的实验室研究并不能验证失效风险相关控制措施的有效性,需要通过适宜的动物模型的相应部位制备骨折或骨缺损模型,评价可降解金属产品在适宜的动物体内的降解性能和产品的安全性及有效性。具体试验项目可包含X光评价、血液中元素分析、组织病理分析、micro-CT分析、植入生物力学评价、周围骨组织分析等。
(十)吻合器
吻合器主要用于组织/脏器的切除、闭合。
1.用于实质脏器或血管切割/吻合的吻合器类产品,因常规的实验室研究并不能充分验证吻合器用于人体的操作性能和吻合性能,宜开展动物实验。
2.对于采用了新材料的肠道或皮肤吻合器类产品,若产品性能、吻合钉材质等与已上市产品存在差异性,仅依靠常规的实验室研究和现有数据不足以评价产品安全性和有效性,宜开展动物实验。另外,通过动物实验可确定产品临床相关参数(如组织厚度等),预测产品在人体中使用时可能出现的安全性问题。
(十一)可吸收外科防粘连产品
对于可吸收外科防粘连产品,应实现产品预期的防粘连功能。该功能宜在适当的活体动物模型上进行研究。动物实验中宜尽可能地体现手术方法、特定手术部位、粘连的类型、粘连的评价方式,以及拟在临床应用时的产品使用方法,并观察产品是否能有效降低粘连的发生率、广泛程度及严重程度等。另外,通过动物实验也可以更好地为临床研究方案设计提供参考。
五、参考资料
[1] General Considerations for Animal Studies for Medical Devices (Draft Guidance)
[2] EFS Guidance: Investigational Device Exemptions (IDEs) for Early Feasibility Medical Device Clinical studies, Including Certain First in Human (FIH) Studies
[3] Recommended Content and Format of Test Reports for Complete Non-Clinical Bench Performance Testing in Premarket Submissions (Draft Guidance)
[4]《超声软组织切割止血系统技术审查指导原则》(国家食品药品监督管理总局通告2018年第37号)
[5]《腔镜用吻合器产品注册技术审查指导原则》(国家食品药品监督管理总局通告2017年第44号)
[6]《腹腔、盆腔外科手术用可吸收防粘连产品注册技术审查指导原则》(国家食品药品监督管理总局通告2016年第7号)
[7]《冠状动脉药物洗脱支架临床前研究指导原则》(国家药品监督管理局通告2018年第21号)
[8]《体外除颤产品注册技术指导原则》(国家食品药品监督管理总局通告2017年第6号)
[9]《腹腔内置疝修补补片动物实验技术审查指导原则》
[10]《钙磷/硅类骨填充材料注册技术审查指导原则》(国家食品药品监督管理总局通告2017年第14号)
[11]《无源植入性医疗器械临床试验审批技术审查指导原则》(国家药品监督管理局通告2018年第40号)
[12]YY/T0316-2016《医疗器械风险管理对医疗器械的应用》
[13]ISO 14971:2007 Medical devices—Application of risk management to medical devices
[14] “Non-Animal Approaches - The Way Forward”, European Commission Scientific Conference summary report, Mark Cronin, March 2017
[15] Directive 2010/63/EU of The European Parliament And of The Council on the protection of animals used for scientific purposes
六、编写单位:
本指导原则由国家药品监督管理局医疗器械技术审评中心编写并负责解释。